The General Linear Model module performs a GLM analysis of the time-series of input images, and is capable of sophisticated analysis of (amongst other things) fMRI data and arterial spin labelling perfusion data. The GLM assumes that the signal changes you see in the time-series consists of linear combination of a set of 'basis' functions. The GLM estimates the amplitudes (weights) of the basis functions such that when they are added together, they form a least-squares fit to the time-series.
You can choose the General Linear Model module by clicking the tab in the Dynamic Analysis tool.
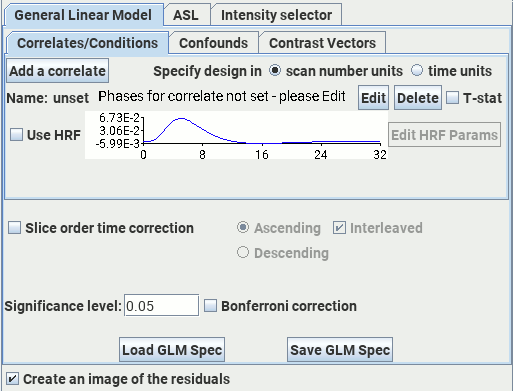
The General Linear Model tab contains three of its own tabs: "Correlates/Conditions", "Confounds" and "Contrast Vectors". These are described below.
 button. To delete a correlate, click on the
button. To delete a correlate, click on the
 button for that correlate.
button for that correlate.
Before creating correlates, decide whether you want to construct your correlates as functions of time or scan number. Select either:
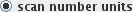 times of onset etc. will be specified with
respect to the scan number; or
times of onset etc. will be specified with
respect to the scan number; or
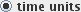 times of onset etc. will be specified in
seconds from the start of the scanning.
times of onset etc. will be specified in
seconds from the start of the scanning.
For each correlate that you have, you will need to construct the basis function for that
correlate. To construct the basis function, click the  button. This will bring up a dialog asking how you want to construct the correlate:
button. This will bring up a dialog asking how you want to construct the correlate:
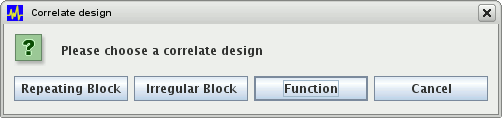
Select one of:
 if the thing you are correlating to
repeats in a regular on-off pattern, such as in a regular block-design fMRI
experiment. Selecting "Repeating Block" brings up the repeating block design dialog:
if the thing you are correlating to
repeats in a regular on-off pattern, such as in a regular block-design fMRI
experiment. Selecting "Repeating Block" brings up the repeating block design dialog:
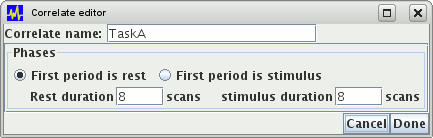
You need to:
 ) or a period of stimulus
(
) or a period of stimulus
( ).
or
.
).
or
.
Click the  button when you are finished, and a
sketch of the design matrix will be shown in the General Linear Model tab for the correlate you
just created.
button when you are finished, and a
sketch of the design matrix will be shown in the General Linear Model tab for the correlate you
just created.
 if the thing you are correlating to
repeats in a irregular on-off pattern, such as in an irregular block-design fMRI experiment.
Selecting "Irregular Block" brings up the irregular block design dialog:
if the thing you are correlating to
repeats in a irregular on-off pattern, such as in an irregular block-design fMRI experiment.
Selecting "Irregular Block" brings up the irregular block design dialog:
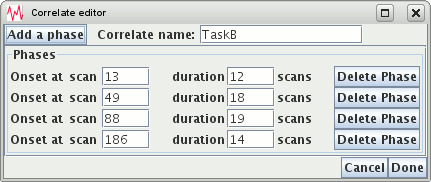
An irregular block design consists of a number (1 or more) phases. One phase consists of a period or rest, followed by a period of stimulus, followed by a period of rest. You can have as many phases are you need, but the phases for one irregular block correlate may not overlap. You need to:
 button.
or
.
button.
or
.
Above, we have set a correlate called "TaskB" that consists of 4 phases with different onset times and durations. The different phases may not overlap; if they do you will see an error message.
Click the button when you are finished, and a
sketch of the design matrix will be shown in the General Linear Model tab for the correlate you
just created.
 if the thing you are correlating to
has a varying amplitude and timing, and you can specify these as analytical functions.
Selecting "Function" brings up the function design dialog:
if the thing you are correlating to
has a varying amplitude and timing, and you can specify these as analytical functions.
Selecting "Function" brings up the function design dialog:
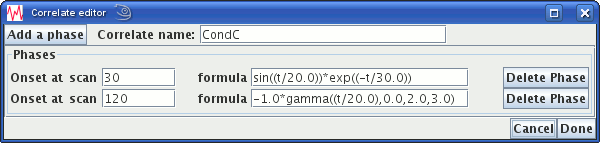
An function correlate design consists of a number (1 or more) functions from which the design matrix is calculated as the sum of all the functions. One function is specified per phase. Of course, you can simply have a singe phase which consists of all the functions you want to use added together, but you may find it simpler and easier to manage if you use a separate phase for each function.
You need to:
button.
or
, the time variable
(t) will be measured either in scan units or seconds.
Above, we have set a correlate called "CondC" that uses two functions: the first is an exponentially-decaying sinusoid, and the second is an inverted gamma probability density function.
Click the button
when you are finished, and a sketch of the design matrix will be shown in the General Linear
Model tab for the correlate you just created.
For each correlate that you set, you will see a sketch of the design matrix next to the correlate name, as shown below for the three example correlates above.
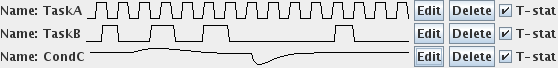
Note: these sketches are only to help you to be sure that you have created your correlates correctly; they are not correctly scaled vertically relative to one another.
Next to each correlate is the  check-box. If selected, a T-static image for the corelate will be produced; if you do not want
T-statistics for any particular correlate, then un-check this box.
check-box. If selected, a T-static image for the corelate will be produced; if you do not want
T-statistics for any particular correlate, then un-check this box.
For functional MRI (fMRI) analysis you can use an HRF by selecting the
 check-box.
The design matrix you have set is convolved with the HRF to give the design matrix that is
actually used in the GLM analysis.
The HRF is defined using two gamma probability density functions, the standard parameters for
which are similar to those used by SPM. However, you can change them by
clicking on the
check-box.
The design matrix you have set is convolved with the HRF to give the design matrix that is
actually used in the GLM analysis.
The HRF is defined using two gamma probability density functions, the standard parameters for
which are similar to those used by SPM. However, you can change them by
clicking on the
 button, which brings up the HRF editor dialog:
button, which brings up the HRF editor dialog:
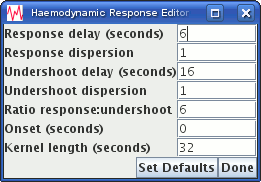
If you change any of the HRF parameters, the HRF shape shown plotted in the HRF display panel will be updated.

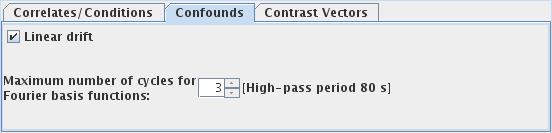
You can model and remove from you data:
 .
.
 spinner, and (as long as you have set the
time between images and the number of time points correctly) Jim will calculate
the equivalent high-pass filter period.
spinner, and (as long as you have set the
time between images and the number of time points correctly) Jim will calculate
the equivalent high-pass filter period.
Note: you can have zero or more correlates. However, you must have fewer correlates and confounds (see below) than you have time points in your data. If you have no correlates, then only the confounds will be used in the GLM; you may wish to do this if you only want to remove confounds from your data, leaving just the residuals.
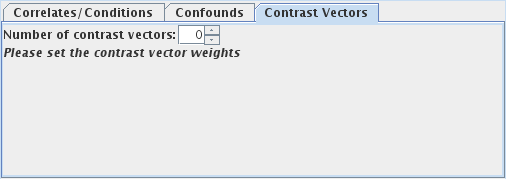
Select the number of contrast vectors you want to calculate, using
the  spinner. This will create a set of
weights for each contrast vector, whereby you can weight the correlate for each task. For example,
imagine you wanted to look at the difference between "TaskA" and "TaskB", and also between "TaskB"
and "CondC" in the example above.
spinner. This will create a set of
weights for each contrast vector, whereby you can weight the correlate for each task. For example,
imagine you wanted to look at the difference between "TaskA" and "TaskB", and also between "TaskB"
and "CondC" in the example above.
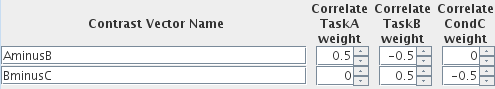
We have set the weights such that we will be looking at the differences between the correlates, and given these contrasts names to reflect those differences. You can use any names for the contrast vectors you wish.
When the GLM analysis is performed, additional SPMs will be produced (two for each contrast vector) - a T-statistic map and a p-value map. The names of the maps will be created using the names you set here.

When you collect data for an MRI scan, the individual slices are normally acquired at different
times. The order of slice collection may vary, starting with the first slice in the dataset, or
the last slice, or the order may be interleaved (1,3,5,7, ...2,4,6,8, ...). You can correct for
the slight time difference in the collection of the slices by selecting
 . Then set whether the slice order is:
. Then set whether the slice order is:
 if the first slice was collected
first and the last slice was collected last (in time).
if the first slice was collected
first and the last slice was collected last (in time).
 if the last slice was collected
first and the first slice was collected last (in time).
if the last slice was collected
first and the first slice was collected last (in time).
 if the slices were collected in
the order 1,3,5,7, ... 2,4,6,8 ... (for ascending order), or N,N-2,N-4,N-6 ... N-1,N-3,N-5,N-7
(for descending order in an N-slice dataset).
if the slices were collected in
the order 1,3,5,7, ... 2,4,6,8 ... (for ascending order), or N,N-2,N-4,N-6 ... N-1,N-3,N-5,N-7
(for descending order in an N-slice dataset).
The GLM module will produce T-statistic and p-value SPMs. You can set the significance level for
these maps by altering the 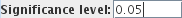 ; pixels where
the statistic is below the significance level is zero will be given an T-value of zero or a
p-value of 1.0 in the corresponding SPMs.
; pixels where
the statistic is below the significance level is zero will be given an T-value of zero or a
p-value of 1.0 in the corresponding SPMs.
Because, for typical images, many thousands of statistical tests are being performed, it is likely
that many pixels will show significance in the SPMs just by chance at a significance level of
0.05 (the default). However, you can perform a Bonferroni correction by selecting the
 check-box. The p-value is adjusted by
taking the number set as the significance level and dividing it by the number of pixels that are
tested using the GLM. Only those pixels that remain significant are output in the SPMs.
check-box. The p-value is adjusted by
taking the number set as the significance level and dividing it by the number of pixels that are
tested using the GLM. Only those pixels that remain significant are output in the SPMs.
 check-box will create
output image(s) with the same names as the input image(s), but with the suffix "residuals". The
residuals image(s) will be the result of subtracting the GLM from the input data. Residuals images
can be useful for investigating whether fitting the GLM still leaves some "structure" - this would
be the case if the GLM does not provide a compete description of the input images, and that
possibly further correlates should be included in the model. In the case of a "complete" GLM, the
residuals will consist entirely of Gaussian-distributed uncorrelated values.
check-box will create
output image(s) with the same names as the input image(s), but with the suffix "residuals". The
residuals image(s) will be the result of subtracting the GLM from the input data. Residuals images
can be useful for investigating whether fitting the GLM still leaves some "structure" - this would
be the case if the GLM does not provide a compete description of the input images, and that
possibly further correlates should be included in the model. In the case of a "complete" GLM, the
residuals will consist entirely of Gaussian-distributed uncorrelated values.
 button, the output images
created will have names produced by taking the base name in the image output section and appending the suffixes from
the correlate and contrast vector names you have provided. The p-value maps are also appended with
"-p" and the T-statistic maps are appended with "-T".
button, the output images
created will have names produced by taking the base name in the image output section and appending the suffixes from
the correlate and contrast vector names you have provided. The p-value maps are also appended with
"-p" and the T-statistic maps are appended with "-T".
When calculating statistical maps, the GLM analysis uses a two-pass analysis. The first fits the GLM to the data to produce a set of residuals that are, in general, auto-correlated. The auto-correlation is then estimated from the residuals, and a second pass corrects the p- and T-values for the auto-correlation in the data. Correction of the SPMs for auto-correlation follows the method of Woolrich et al..
 button. This will bring up a
File Chooser for you to select the XML file name.
button. This will bring up a
File Chooser for you to select the XML file name.
Clicking on the  button will allow you to
select an XML file where you previously save the setup, and reload it from disk.
button will allow you to
select an XML file where you previously save the setup, and reload it from disk.
The document type definition for a GLM specification file is given in the file formats section.